Dr Antoine MONSEL
Réanimation Chirurgicale Polyvalente (Pr LANGERON)
Département d’Anesthésie – Réanimation Chirurgicale (Pr PUYBASSET)
Hôpital Universitaire Pitié-Salpêtrière – Charles Foix, Paris 6
Tél: +33(0)1.84.82.73.99 – Fax: +33(0)1.42.17.58.79
Sorbonne Universités, Université Pierre et Marie Curie
INSERM – UMR S 959 – Immunologie-Immunopathologie-Immunothérapie (i3)
Le SDRA est un œdème pulmonaire dit « lésionnel », c’est à dire entraîné par une hausse de la perméabilité capillaire pulmonaire survenant à la suite d’une agression directe ou indirecte de la membrane alvéolo-capillaire, associé à une inflammation pulmonaire intense et une hypoxémie sévère. Sa mortalité varie de 30 à 45% en fonction des études.
Sa définition a été réévaluée récemment lors de la conférence de Berlin (Force, et al. 2012). Elle repose sur 4 critères : 1) insuffisance respiratoire aiguë évoluant depuis une semaine ou moins, 2) opacités bilatérales sur l’imagerie thoracique, 3) pas d’arguments pour un œdème hydrostatique prédominant, 4) hypoxémie avec rapport PaO2/FIO2 < 300 mmHg pour une pression télé-expiratoire positive réglée à 5 cmH2O ou plus avec 3 stades de gravité définis en fonction de l’hypoxémie.
La prise en charge est centrée sur le diagnostic, le traitement étiologique et l’assistance ventilatoire surtout invasive. Les réglages du ventilateur doivent éviter d’induire des lésions pulmonaires supplémentaires qui contribuent à obérer le pronostic du malade. Il est essentiel de limiter les volumes et les pressions pulmonaires. Une curarisation précoce et courte (48 heures) ainsi que la mise en décubitus ventral précoce dans les formes les plus sévères réduit la mortalité. Maintenir un bilan hydrosodé négatif, une fois le choc contrôlé, permet de réduire la durée globale de ventilation mécanique. Une corticothérapie peut être mise en route en cas de SDRA non-résolutif. De nombreuses évaluations sont en cours notamment en ce qui concerne les techniques d’assistance respiratoire extracorporelle soit pour améliorer l’oxygénation soit pour épurer le gaz carbonique.
1. Définitions
Historiquement, le SDRA a été défini en 1967 par une triade cardinale (hypoxémie réfractaire, infiltrats alvéolo-interstitiels bilatéraux, absence d’insuffisance cardiaque gauche congestive au premier plan), survenant chez des soldats américains au cours de la guerre du Viêt-Nam. Victimes d’un polytraumatisme ouvert à la suite de plaies par balles, opérés en urgence et transfusés, ils développaient 2 à 3 jours plus tard un tableau de détresse respiratoire aiguë avec hypoxémie intense, réfractaire aux hauts débits d’oxygène pur, et opacités alvéolaires bilatérales sur la radiographie pulmonaire. Il s’agit donc d’une affection caractérisée par une altération grave des échanges gazeux, la présence d’infiltrats alvéolaires et interstitiels (figure 1, B et C), sans que l’œdème pulmonaire qui accompagne cette affection ne soit dû à une défaillance ventriculaire gauche (Ashbaugh, et al. 1967, Bernard, et al. 1994). L’examen histologique des poumons montre la présence d’un exsudat riche en polynucléaires neutrophiles (PMN), avec des zones hémorragiques, témoignant d’une destruction de la barrière alvéolocapillaire (figure 1, A, et 2) (Ware and Matthay 2000).
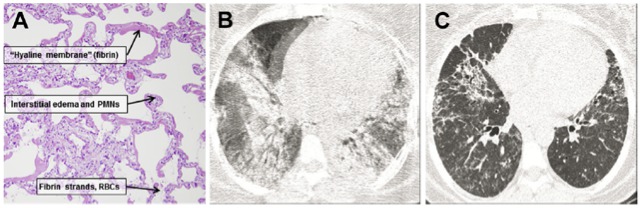
Figure‑1: Caractéristiques histologiques et scannographiques du SDRA. (A) Coloration par hématoxyline-éosine d’un échantillon de biopsie pulmonaire d’un patient présentant un SDRA. On peut constater la présence de membranes hyalines tapissant la surface alvéolaire, d’un œdème interstitiel étendu et de PMN. L’espace alvéolaire contient également des hématies et des dépôts de fibrine. Les coupes scannographiques B et C représentent les lésions pulmonaires rencontrées lors des phases exsudatives (B) et fibro-prolifératives (C) du SDRA. La phase exsudative est caractérisée par des images en verre dépoli et des consolidations alvéolaires, tandis que la phase fibro-proliférative par des opacités linéaires persistantes, des bronchectasies en traction et des nodules en nid d’abeille. SDRA = syndrome de détresse respiratoire aiguë, PNN= polymorphonucléaires neutrophiles. D’après (Standiford and Ward 2016).
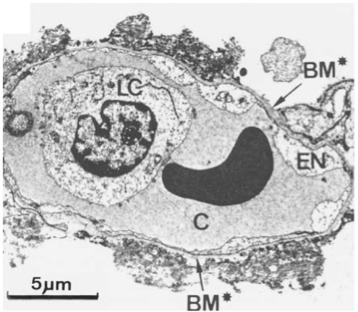
Figure ‑2 : Image de microscopie électronique de l’unité alvéolo-capillaire durant le SDRA. Des lésions sont présentes sur le versant endothélial du capillaire pulmonaire ainsi que sur le versant épithélial de l’alvéole. LC représente un PNN intravasculaire au sein du capillaire pulmonaire (C). La vacuolisation et l’œdème du capillaire sont visibles. La perte du contingent cellulaire épithélial avec la formation de membranes hyalines sur le versant épithélial de la lame basale sont également présents (BM*). SDRA = syndrome de détresse respiratoire de aiguë. D’après (Ware and Matthay 2000).
Depuis, plusieurs définitions ont été proposées dont aucune n’est vraiment satisfaisante car n’appréhendant pas réellement l’hétérogénéité (c’est un syndrome) et la complexité de la physiopathologie du SDRA. La définition de l’American-European Consensus Confernce de 1994 souffrait d’une spécificité médiocre, d’une mauvaise corrélation pronostique, ainsi que de la nécessité de recourir à un cathétérisme cardiaque droit tombé en désuétude depuis les années 2000. La définition la plus récente (tableau 1) propose de définir le SDRA par l’association des 4 critères ci-dessous (Force, et al. 2012) :
- Insuffisance respiratoire aiguë qui évolue depuis une semaine ou moins
- Opacités bilatérales visibles sur l’imagerie thoracique
- Œdème pulmonaire dont la participation hydrostatique n’est pas prédominante.
- Hypoxémie définie à partir du rapport PaO2/FIO2, où FIO2 est la fraction inspirée d’oxygène dans l’air insufflé par le respirateur qui permet de classer le SDRA en 3 stades :
- Léger : 200<PaO2/FIO2 ≤ 300 mmHg
- Modéré : 100<PaO2/FIO2≤ 200 mmHg
- Sévère : PaO2/FIO2≤100 mmHg
2. Incidence et pronostic
Incidence
Le SDRA concerne encore aujourd’hui 6 à 58 cas sur 100 000 habitants chaque année dans le monde (Bersten, et al. 2002, Brun-Buisson, et al. 2004, Caser, et al. 2014, Esteban, et al. 2002, Herridge, et al. 2003, Herridge, et al. 2011, Hughes, et al. 2003, Li, et al. 2011, Luhr, et al. 1999, Reynolds, et al. 1998, Rubenfeld, et al. 2005, Wind, et al. 2007) (Tableau 2 de la version longue du texte). Aux Etats-Unis, une étude a mis en évidence une incidence de SDRA de 78,9 cas pour 100 000 personnes par an et de 86,2 cas pour 100 000 personnes par an après ajustement pour l’âge (Rubenfeld, et al. 2005). En réanimation, l’incidence est de 34 cas pour 100 000 personnes par an en Australie (Esteban, et al. 2002) et de 17,9 cas pour 100 000 personnes par an en Scandinavie (Wind, et al. 2007). Dans une étude de cohorte multicentrique Européenne (Brun-Buisson, et al. 2004) qui incluait 6522 patients de réanimation, un SDRA était présent chez 7,1% des patients admis en réanimation. Cette proportion atteignait 12,5% au-delà d’un séjour de 24 heures en réanimation. Une autre étude reportait une incidence du SDRA de 4,5% chez les patients ventilés à leur admission en réanimation (Esteban, et al. 2002).
Pronostic
Bien qu’en baisse, la mortalité du SDRA s’élève actuellement à 30-45% (Bersten, et al. 2002, Brun-Buisson, et al. 2004, Caser, et al. 2014, Herridge, et al. 2003, Herridge, et al. 2011, Hughes, et al. 2003, Li, et al. 2011, Luhr, et al. 1999, Pierrakos and Vincent 2012, Rubenfeld, et al. 2005, Wind, et al. 2007) (Tableau 2). Certaines études incluant des patients sélectionnés sur des critères strictes ont récemment décrit des taux de mortalité avoisinant 20%. Cependant dans les études prospectives de cohorte évaluant la mortalité des patients en SDRA sans sélection préalable, des taux de mortalité variant de 27% à 45% pouvant même atteindre 70% selon les comorbidités et la sévérité du SDRA, sont classiquement rapportés (Brun-Buisson, et al. 2004, Esteban, et al. 2002, Pierrakos and Vincent 2012, Reynolds, et al. 1998, Rubenfeld, et al. 2005). L’âge (> 60 ans), la présence de comorbidités préexistantes, la sévérité de l’hypoxémie (PaO2/FiO2 < 100), l’augmentation de l’espace mort alvéolaire (> 0,60), le choc septique et les défaillances d’organes associées augmentent la mortalité du SDRA (Matthay, et al. 2012).
La défaillance multiviscérale est la cause de décès la plus représentée, alors que la période des 7-10 premiers jours semble déterminante en terme de pronostic des patients. Les jeunes patients polytraumatisés présentent le meilleur pronostic avec une récupération fonctionnelle respiratoire ad integrum en 6 à 12 mois. Des altérations modérées obstructives et/ou restrictives peuvent persister chez certains patients (Herridge, et al. 2003, Herridge, et al. 2011).
3. Etiologies
Atteinte pulmonaire directe ou indirecte.
Le SDRA est l’expression d’une agression de la membrane alvéolocapillaire qui peut être directe, sur le versant épithélial de la membrane, comme au cours des pneumonies infectieuses, ou indirecte, portant sur le versant endothélial comme à la suite d’un choc septique, ou bien d’une agression inflammatoire non septique d’origine extra-pulmonaire (tableau 3). Le sepsis est ainsi la première cause de SDRA.
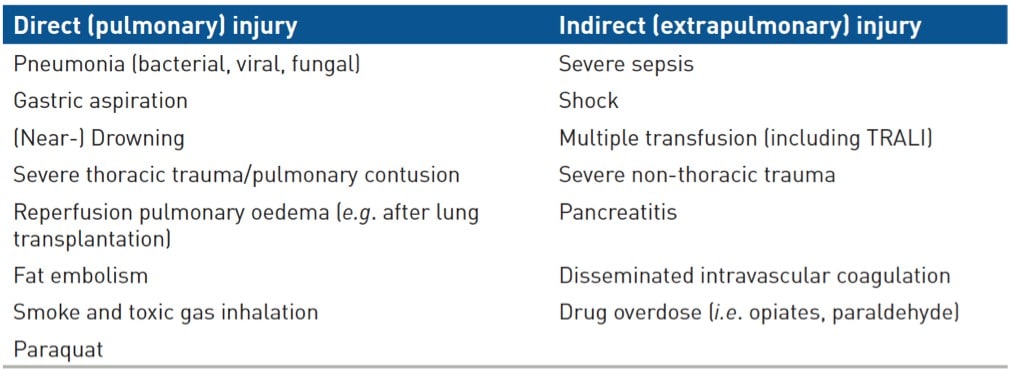
Tableau 3: Principales étiologies de SDRA. SDRA = syndrome de détresse respiratoire aiguë, TRALI = transfusion-related acute lung injury. D’après (Ware and Matthay 2000)
Facteurs environnementaux et génétiques
L’éthylisme chronique, le tabagisme actif et passif, constituent des facteurs environnementaux augmentant le risque de développer un SDRA en cas d’agression pulmonaire, via des effets sensibilisateurs de l’épithélium, de l’endothélium et des autres cellules immunitaires de la membrane alvéolocapillaire (Matthay, et al. 2012). La présence d’un sepsis, d’une maladie pulmonaire chronique et d’un pH plasmatique abaissé ont également été montrés comme des facteurs précipitants la survenue de SDRA (Matthay, et al. 2012). D’autres études menées dans des populations indépendantes devront être menées, avant de conclure formellement à l’identification de certains gènes relatifs à l’hôte ou la virulence de l’agent pathogène causal, comme favorisants la survenue de SDRA.
4. Physiopathologie
La grande majorité des voies de signalisation convergeant vers la mort cellulaire (nécrose ou apoptose) des cellules clef de l’unité alvéolocapillaire pulmonaire, sont impliquées dans la physiopathologie du SDRA. C’est la détection de motifs moléculaires exogènes provenant d’agents pathogènes ou endogènes issus de tissu lésé de l’organisme, qui constitue l’élément déclencheur initial de l’activation de ces voies de signalisations. Si les macrophages alvéolaires et systémiques orchestrent essentiellement la détection de ces motifs moléculaires, ce sont les polymorphonucléaires neutrophiles (PNN) qui constituent les principaux effecteurs de la réponse inflammatoire dérégulée conduisant à la destruction de l’unité alvéolocapillaire du SDRA.
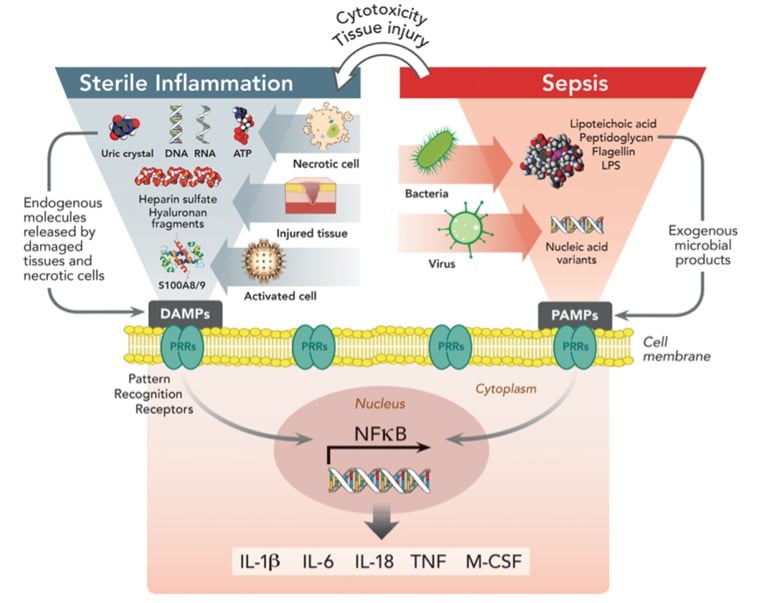
Figure 3: Pattern recognition receptors (PRRs) et leur implication dans l’inflammation d’origine infectieuse ou stérile. Les PRRs qui peuvent être soit membranaires ou intra-cytoplasmiques (NLRs pour nucleotide-binding oligomerization domain-like receptors), sont exprimés par les cellules de l’immunité innée (monocytes, macrophages et cellules dendritiques) mais également par les cellules épithéliales et endothéliales constituant l’unité alvéolocapillaire. Les récepteurs de la superfamille Toll-like recpetors (TLRs) constituent une catégorie majeure de PRRs membranaires particulièrement conservée parmi les vertébrés. L’interaction entre PAMPs, DAMPs et PRRs est à l’origine de la transduction d’un signal d’activation des voies de signalisation intracellulaires aboutissant notamment à l’activation de facteur de transcription nucléaire kappa B (NF-kB). Ce facteur de transcription est transloqué en intra-nucléaire et est responsable de l’activation de la transcription de plusieurs gènes codant pour la synthèse de protéines aiguës de la phase inflammatoire. Des quantités importantes d’IL-1β, IL-6, IL-8, de TNF-α et M-CSF sont donc produites et sécrétées par les cellules activées, c’est à dire majoritairement les macrophages alvéolaires, les cellules épithéliales et endothéliales de l’unité alvéolocapillaire pulmonaire. PRRs = pattern recognition receptors; PAMPs = pathogen-associated molecular patterns; DAMPs = damage-associated molecular patterns; LPS = lipopolysaccharide; ATP = adenosine triphosphate; NF-κB = nuclear factor kappa-light-chain-enhancer of activated B cells; IL-1β = interleukin-1 beta; IL-6 = interleukin 6; IL-18 = interleukin 18; TNF = tumor necrosis factors; M-CSF = macrophage colony-stimulating factor. D’après (Monsel, et al. 2014)
Rôles des polynucléaires neutrophiles dans le SDRA
L’activation des cellules de premières lignes de l’immunité innée présentes au sein de l’unité alvéolocapillaire agressée, c’est à dire les macrophages et PNN, conduit à la libération de quantité importante de chimiokines à l’origine d’un chimiotactisme puissant des PNN circulants. L’IL-8 (CXCL8) semble jouer un rôle déterminant dans l’attraction massive des PNN circulants au sein de l’espace alvéolaire pulmonaire (Han and Mallampalli 2015).
Une fois dans l’alvéole pulmonaire, les PNN jouent un rôle crucial dans l’induction de l’inflammation dérégulée du SDRA (Figure 4). L’importance de leur infiltrat alvéolaire est un facteur de mauvais pronostic et est corrélée avec la sévérité du syndrome. Le rôle clef des PNN en tant que premier effecteur pro-inflammatoire réside dans plusieurs mécanismes :
-sécrétions de cytokines pro-inflammatoires, de protéases et de leukotriènes induisant des lésions épithéliales et endothéliales
-sécrétions de cytokines pro-inflammatoires induisant l’activation des cellules clef de l’immunité innée et adaptative
-activation du système plaquettaire et de coagulation
-induction de stress oxydatif via l’explosion oxydative ou burst oxydatif
-induction de lésions cellulaires et tissulaires via le processus de nétose
-induction de lésions cellulaires et tissulaires via la sécrétion d’histones
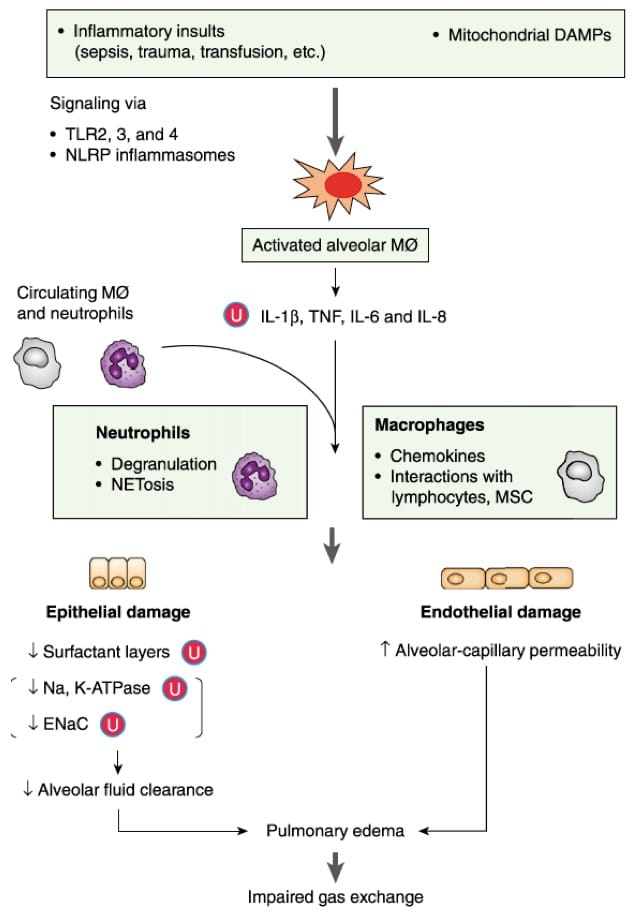
Figure 4: Immunopathologie et physiopathologie du SDRA. Les motifs provenant des lésions initiales soit de pathogènes (PAMPs) soit d’alarmines libérées lors de lésions cellulaires (DAMPs), incluant les DAMPs d’origine mitochondriales, activent les TLR et NLR des macrophages alvéaolaires. Les macrophages activés libèrent des cytokines pro-inflammatoires et recrutent massivement via les chémokines des PNN et des macrophages circulants vers les alvéoles lésées. L’afflux massif non contrôlé de PNN et l’activation persistante non régulée des macrophages et des PNN alvéolaires sont à l’origine de lésions extensives de la barrière alvéolo-capillaire. La rupture de cette barrière génère un œdème alvéolaire riche en protéine qui conduit à l’altération marquée des échanges gazeux alvéolo-capillaires. L’ubiquitination ((U)) joue un rôle important dans la régulation de protéines clef dans le SDRA, aboutissant à la sécrétion de cytokines, la réduction des protéines du surfactant et à l’altération de la fonction des échangeurs ioniques. Le SDRA est associé à la déplétion en surfactant, conduisant à l’augmentation du phénomène de Nétose, un mécanisme qui participe à la destruction des cellules clef de l’unité alvéolo-capillaire. Enfin, les DAMPs d’origine mitochondriale peuvent augmenter la perméabilité endothéliale microvasculaire indépendamment des leucocytes. DAMPs = danger-associated molecular patterns; ENaC = epithelial sodium chanel; IL = interleukin; MSC = mesenchymal stem cell; NETosis = neutrophil extracellular traps; NLRP = NOD-like receptors; PAMPs = pathogen-associated molecular patterns; TLR = toll-like receptors; TNF = tumor necrosis factor.D’après (Han and Mallampalli 2015)
Parallèlement à cette polarité pro-inflammatoire, majoritairement représentée lors de la phase initiale du SDRA, il est important de préciser que certaines populations de PNN sont également capables de participer à la phase résolutive, plus tardive du SDRA, via la sécrétion de cytokines immunorégulatrices tells que le TGF-β, IL-10 ou IL-1RA, l’induction de leur apoptose, et l’induction de l’expansion et de l’activation d’autres populations immunorégulatrices tels que les lymphocytes T régulateurs (Ortega-Gomez, et al. 2013).
Autres voies de signalisation impliquées dans le SDRA
D’autres voies de signalisation à l’origine du dommage de l’unité alvéolo-capillaire sont également mises en jeu dans le SDRA. Les lésions induites par ischémie-reperfusion sont notamment à l’origine d’une perturbation de l’homéostasie métabolique cellulaire. Une déplétion majeure des enzymes clef du métabolisme énergétique cellulaire a été observée et montrée comme responsable de l’effondrement des stocks en ATP des cellules épithéliales et endothéliales alvéolaires. L’activation des voies de signalisation pro-apoptotiques, et l’induction d’un stress oxydatif ont également été démontrées comme pourvoyeurs de lésions tissulaires induites par ischémie-reperfusion dans le SDRA. L’activation de la voie des médiateurs lipidiques (leukotriènes, lipoxines) ainsi que de la voie du complément, de l’activation plaquettaire et de la coagulation sont également mis en jeu dans la genèse des lésions de l’unité alvéolo-capillaire observées dans le SDRA (Han and Mallampalli 2015, Matthay, et al. 2012).
Concept de volotrauma
Le concept de volotrauma est venu compléter la compréhension des processus physiopathologiques à l’origine des lésions de l’unité alvéolo-capillaire rencontrées dans le SDRA. La ventilation protectrice, c’est à dire avec l’administration de faibles volumes courant (6 mL/kg) réduit l’œdème alvéolaire, et protège l’homéostasie épithéliale et endothéliale de la barrière alvéolo-capillaire dans le SDRA. A l’inverse, les forces d’étirement imposées à l’alvéole lors de l’administration de hauts volumes courants exacerbent l’agression pulmonaire. Cette aggravation du syndrome par l’administration de volumes courants trop importants définit le volotrauma (Matthay, et al. 2012). Les marqueurs de lésions épithéliales sont abaissés chez les patients en SDRA bénéficiant d’une ventilation protectrice. Les voies de signalisations inflammatoires dites mécano-sensitives sont ainsi réprimées lors de la ventilation protectrice, aboutissant à la réduction des niveaux plasmatiques et alvéolaires d’IL-6, IL-8, et de récepteur soluble du TNF de type 1 (Figure 5) (Matthay, et al. 2012).
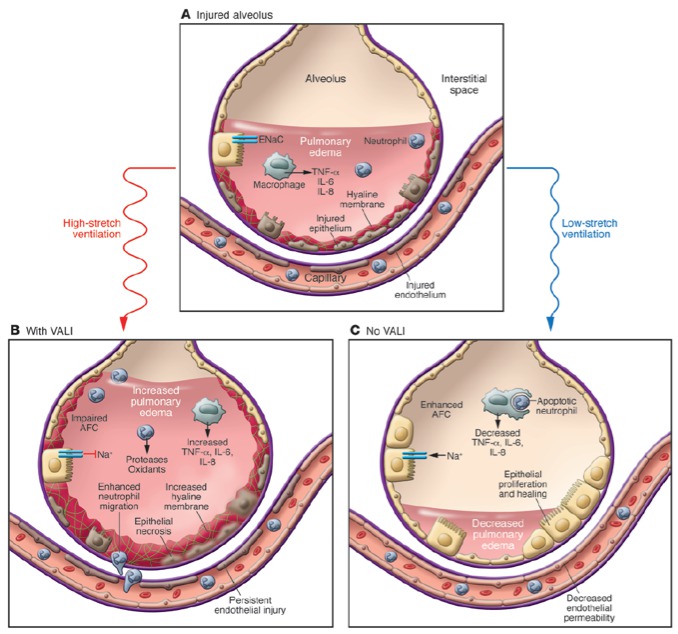
Figure 5: Mécanismes des lésions induites par la ventilation artificielle, ou volotrauma. (A) Les lésions inflammatoires aiguës pulmonaires induisent une activation des macrophages alvéolaires conduisant à la sécrétion massive de cytokines et de chémokines chémoattractives de PMN, qui à leur tour, sécrètent des cytokines pro-inflammatoires qui lèsent les différents acteurs clef de l’unité alvéolo-capillaire. L’augmentation de la perméabilité alvéolo-capillaire aux protéines qui en résulte, conduit à l’inondation alvéolaire d’un œdème inflammatoire riche en protéines, et à la destruction du surfactant. (B) Une ventilation artificielle invasive agressive à hauts volumes et/ou hautes pressions aura pour conséquences une exacerbation des lésions initiales avec une augmentation des lésions de nécrose de l’épithélium et de l’endothélium alvéolaire, de l’infiltrat alvéolaire en PMN, des lésion de stress oxydatifs, de la sécrétion macrophagique en cytokines pro-inflammatoires et des dépôts de fibrine et de membranes hyalines. Ce type de ventilation peut aussi altérer les mécanismes de clairance alvéolaire en eau libre. (C) A l’opposé, une ventilation protectrice peut limiter les lésions de l’unité alvéolo-capillaire, en diminuant la libération de cytokines pro-inflammatoires, et en restaurant une clairance alvéolaire efficace, permettant la mise en place des mécanismes de réparation de l’épithélium et de l’endothélium alvéolaire. Cette réparation se fait par le biais de la migration, la prolifération et la différenciation des pneumocytes de type II qui repeuplent progressivement la membrane basale dénudée. L’apoptose des PNNqui sont ensuite phagocytés par les macrophages alvéolaires est également un des mécanismes essentiels de cette phase résolutive du SDRA. D’après (Matthay, et al. 2012)
Lésions endothéliales et épithéliales
L’infiltrat majeur de PNN pulmonaires et leur activation subsidiaire, génèrent la libération d’une grande quantité de cytokines et de chimiokines pro-inflammatoires conduisant à la mort cellulaire via inflammation, apoptose, stress oxydatif et altération du métabolisme énergétique. L’unité alvéolo-capillaire est particulièrement touchée par cette mort cellulaire avec notamment une perte massive de pneumocytes I et II, de cellules endothéliales et de cellules immunitaires alvéolaires (macrophages, lymphocytes, PMN), ou interstitielles (monocytes-macrophages, lymphocytes, PNN) (Ware 2006). Ces dommages alvéolocapillaires diffus sont à l’origine de la destruction du surfactant, de l’altération de la résorption de l’œdème alvéolaire et de l’augmentation de la perméabilité membranaire aux protéines plasmatiques. L’œdème lésionnel ou inflammatoire consécutif à ces lésions pulmonaire est à l’origine du SDRA (Ware 2006). La déstabilisation des ponts de vascular endothelial (VE)-cadhérines entre les cellules endothéliales a été démontrée comme essentielle dans l’augmentation de la perméabilité endothéliale dans plusieurs modèles de SDRA. Ces ponts sont normalement régulés par les interactions cytoplasmiques entre caténines et actines, par l’organisation du cytosquelette et les voies de signalisations intracellulaires des Rho et Rac kinases. La destruction de ces jonctions cadhérines entraine une fuite capillaire dans l’interstitium pulmonaire mais facilite également la transmigration des PNN normalement très régulée dans le processus de diapédèse (Matthay, et al. 2012) (Figure 6).
Les différentes phases chronologiques du SDRA
Les 3 phases du SDRA classiquement décrites se succèdent selon la séquence histologique suivante :
-phase aiguë inflammatoire de J1 à J7 (infiltrats de PNN, membranes hyalines, hémorragies intra-alvéolaires, figure 7).
–phase subaiguë de J7 à J14 s’organisant avec un début de réparation cellulaire (prolifération des pneumocytes II, infiltration fibroblastiques interstitielle et dépôt de collagène, figure 8).
–phase chronique fibro-proliférative au-delà de J14 (réparation alvéolaire et fibrose interstitielle)
Que ce soit durant la phase aiguë inflammatoire ou la phase plus tardive résolutive, les cellules immunitaires telles que monocytes-macrophages, PNN et lymphocytes jouent donc un rôle déterminant dans la physiopathologie du SDRA. Elles altèrent le fonctionnement de l’épithélium, de l’endothélium et du stroma alvéolo-interstitiel, directement responsables de l’œdème lésionnel. Les traitements immunomodulateurs offrent donc un axe de recherche particulièrement attractif, dont l’objectif est d’équilibrer dans le temps et l’espace la dynamique de la réaction immuno-inflammatoire dans le SDRA. Des thérapeutiques immunomodulatrices capables de pondérer la réaction pro-inflammatoire durant la phase aiguë tout en accentuant le versant pro-résolutif durant la phase subaiguë à tardive, représenteraient un nouvel espoir dans le traitement du SDRA.
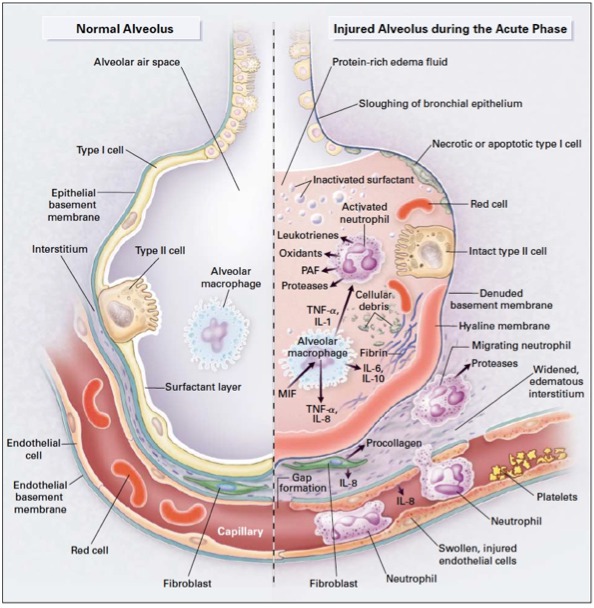
Figure ‑7: Physiopathologie de la phase exsudative précoce du SDRA. L’activation des macrophages alvéolaires par les voies des PRRs via la détection de PAMPs et DAMPs entraine leur activation et la sécrétion de grande quantité de cytokines pro-inflammatoires et de chémokines chemo-attractantes. L’infiltrat massif de PNN entraine alors la sécrétion d’autres cytokines inflammatoires qui lèsent les acteurs principaux de l’unité alvéolocapillaire, à savoir les pneumocytes de type I et II, les cellules endothéliales et les fibroblastes du tissu interstitiel environnant. La rupture de la barrière alvéolo-capillaire entraine l’extravasation d’un liquide exsudatif riche en protéines entrainant une inondation alvéolaire. Des micro-thrombi se forment également au sein des capillaires. Le surfactant est détruit et un dépôt de membranes hyalines se constitue. D’après (Ware and Matthay 2000)
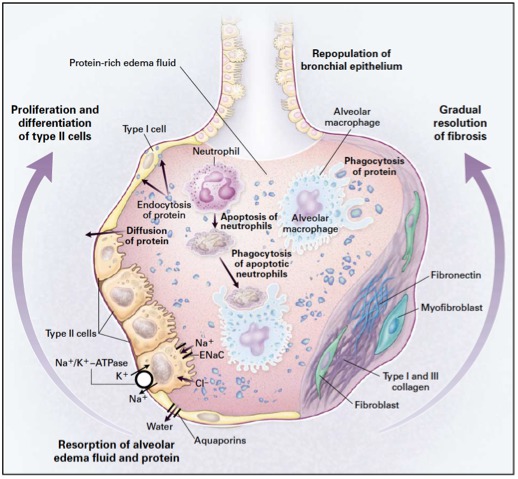
Figure 8: Physiopathologie des phases résolutive et fibro-proliférative tardive du SDRA. A partir de la deuxième semaine, et si l’étiologie du SDRA a été contrôlée, dans le meilleur des cas, une phase résolutive s’instaure. Elle est caractérisée par une activation de la migration, de la prolifération et de la différenciation des pneumocytes de type II en pneumocytes de type I, afin de repeupler la membrane basale alvéolaire totalement dénudée au décours de la lésion inflammatoire initiale. Ce repeuplement restaure peu à peu la clairance alvéolaire permettant une clairance progressive de l’œdème alvéolaire. Les PNN en excès meurent par apoptose et sont phagocytés par les macrophages alvéolaires. Ces derniers éliminent également les membranes hyalines et autres débris cellulaires au sein de l’alvéole en voie de réparation. Dans une minorité des cas, cette phase résolutive peut, si elle n’est pas contrôlée, aboutir à une fibro-prolifération intense de fibroblastes pulmonaires, aboutissant à un stade de fibrose pulmonaire organisée. D’après (Ware and Matthay 2000)
5. Biomarqueurs
Il est actuellement impossible de ne pas aborder la question des biomarqueurs lorsqu’il s’agit de s’intéresser au SDRA. En effet, depuis 20 ans, les progrès techniques en terme de capacité de gestion et d’analyse de lourde série de données biocliniques ont permis d’aborder le SDRA par un versant biologique moléculaire. Une revue de la littérature publiée récemment synthétise l’ensemble des biomarqueurs protéiques ayant démontré une expression différentielle dans le SDRA, durant la phase aiguë et résolutive (Blondonnet, et al. 2016) (Figure 9).
Le développement de stratégies d’analyses “multi-omics” (Rogers and Matthay 2014) exploitant des données génomiques, transcriptomiques, protéomiques et métabolomiques, permet d’appréhender le syndrome dans les différentes étapes d’expression moléculaire, de l’amont (génome) vers l’aval (réponse métabolomique) (Figure 10).
La finalité de l’étude des biomarqueurs dans le SDRA est triple. Elle permettra à terme de:
-progresser dans la compréhension des mécanismes physiopathologiques du syndrome
-dépister les patients à risque permettant la mise en œuvre de stratégies préventives
-mettre en évidence de nouvelles cibles thérapeutiques potentielles
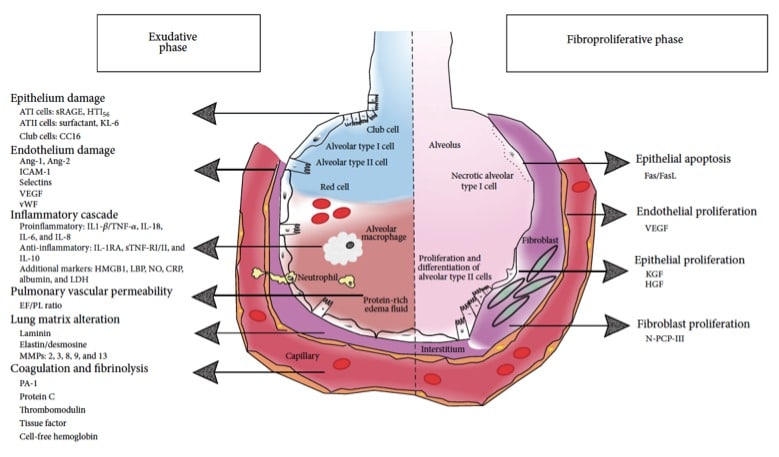
Figure 9: Principaux biomarqueurs décrits présents dans les phases précoces et tardives du SDRA. Ang-1 = angiopoietin-1 ; Ang 2 = angiopoietin-2; ATI = alveolar type I cells; ATII = alveolar type II cells; CRP = C-reactive protein; EF-PL ratio = pulmonary edema fluid-to-plasma protein ratio; HMGB1 = High mobility group box nuclear protein 1; HGF = hepatocyte growth factor; KGF = keratinocyte growth factor; sICAM = soluble intercellular adhesion molecule-1; IL = interleukin; LBP = Lipopolysaccharide binding protein; LDH = lactate deshydrogenase; MMP = matrix metalloproteinases; NO = nitric oxide; N-PCP-III = Pulmonary fibroblasts produce procollagen III peptide; PAI-1 = plasminogen activator inhibitor-1; sRAGE = soluble form of receptor for advanced glycation endproducts; TNF = tumor necrosis factor; VEGF = vascular endothelial growth factor; vWF = von willebrand factor ; D’après (Blondonnet, et al. 2016)
6. Traitements
Les principales grandes études cliniques ayant testé des thérapeutiques dans le SDRA sont synthétisées et discutées dans une revue de la littérature (Matthay, et al. 2012) et représentées dans le Tableau 4.
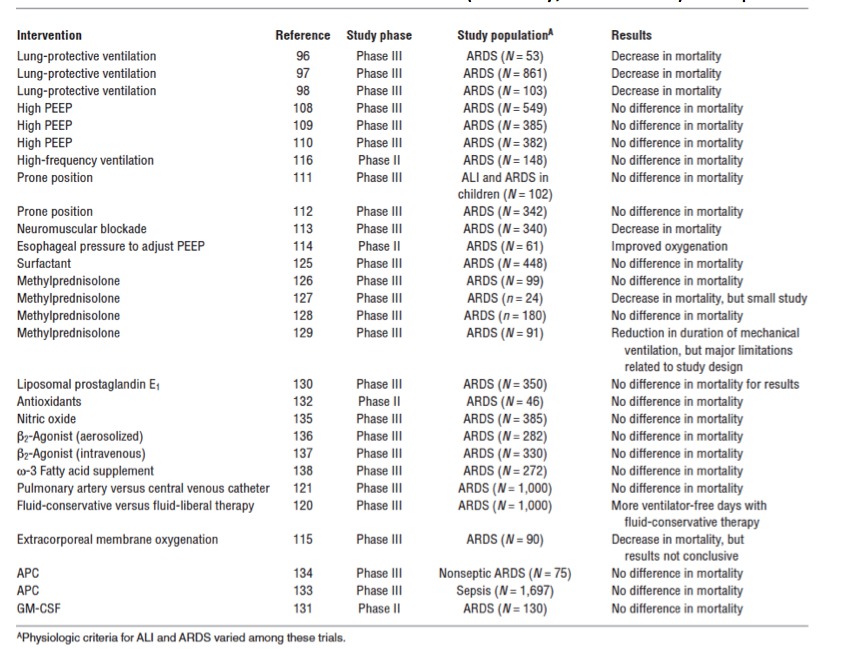
Tableau 4: Sélection d’une série d’études princeps ayant testé des thérapeutiques dans le SDRA. D’après (Matthay, et al. 2012)
Gestion de la ventilation artificielle
Les 2 objectifs principaux de la ventilation mécanique conventionnelle au cours du SDRA actuellement sont de maintenir une oxygénation correcte (objectif de PaO2 entre 55 et 80 mmHg) et de prévenir le volotrauma (concept de ventilation protectrice). Les réglages du ventilateur privilégient un faible volume courant (6 mL/kg de poids idéal), une pression expiratoire positive optimisée et la surveillance de la pression alvéolaire (pression de plateau) à maintenir en-dessous de 28-30 cmH2O. Cette stratégie réduit la mortalité de 9% par rapport à des volumes courants plus élevés (2000, Amato, et al. 1998, Villar, et al. 2006) (Tableau 4). L’utilisation de faibles volumes courants peut générer une hypoventilation alvéolaire avec acidose respiratoire qui est à respecter jusqu’à une limite de pH plasmatique de 7,20-7,30 (en dehors du contexte de trauma crânien).
Le décubitus ventral améliore l’oxygénation sanguine par redistribution de la ventilation pulmonaire vers les zones dorsales alors que la perfusion reste prédominante dans ces régions, homogénéise les contraintes mécaniques au sein du poumon et réduit les lésions de volotrauma. Au cours du SDRA sévère, le DV réduit la mortalité lorsqu’il est pratiqué précocément chez les patients les plus sévères (P/F < 200) (Guerin, et al. 2013).
Bilan hydrosodé
Un large essai randomisé a montré que le maintien d’un bilan hydrosodé négatif grâce à un apport restrictif en solutés de remplissage, une fois contrôlée l’insuffisance circulatoire initiale permet d’augmenter le nombre de jours vivants sans ventilation mécanique de plus de 2 jours par rapport à une politique libérale d’administration des fluides La mortalité n’est toutefois pas modifiée significativement (Kuebler, et al. 1999, National Heart, et al. 2006, National Heart, et al. 2006) (Tableau 4).
Dans un essai de petite taille chez des malades avec SDRA et hypo protidémie, la perfusion d’albumine associée à un traitement diurétique permet d’améliorer l’oxygénation et de maintenir le bilan hydrosodé négatif.
Traitements pharmacologiques
Un déficit quantitatif et/ou fonctionnel d’un ou plusieurs composants du surfactant endogène a été régulièrement mis en évidence dans le LBA de malades avec SDRA. L’apport exogène de surfactant, naturel ou de synthèse, a été réalisé chez des malades avec SDRA dans de nombreuses études, par voie inhalée ou par bronchoscopie, sans aucun bénéfice démontré (Spragg, et al. 2004)(Tableau 4).
En comparant des malades avec SDRA traités par sédation et curarisation pendant 48 heures à un groupe n’ayant reçu que la sédation pendant la même période, il a été démontré: 1) une amélioration de l’oxygénation, 2) une réduction de l’inflammation pulmonaire et systémique et 3) une réduction de mortalité dans le groupe sédation et curares par rapport au groupe de contrôle (sédation et placebo). Les mécanismes d’action évoqués sont la réduction des lésions induites par volotrauma par réduction de la pression trans-pulmonaire régionale et/ou un effet anti-inflammatoire propre de la molécule (Papazian, et al. 2010)(Tableau 4).
La question thérapeutique des corticoïdes est encore très débattue. Un large essai n’a pas montré d’effet bénéfique des corticoïdes administrés systématiquement au stade de SDRA non résolutif par rapport au placebo. Un début tardif des corticoïdes, après la deuxième semaine d’évolution, était même délétère dans cette étude. En pratique, devant des critères de SDRA persistant à J7-J10 après avoir éliminé d’autres causes d’hypoxémie une corticothérapie peut être discutée (Bernard, et al. 1987, Meduri, et al. 2007, Meduri, et al. 1998, Steinberg, et al. 2006) (Tableau 4). La place de la biopsie pulmonaire chirurgicale pour décider de débuter les corticoïdes n’a, pour l’instant, pas été évaluée.
La résorption de l’œdème alvéolaire passe par la mise en jeu des récepteurs β2-adrénergique au niveau de l’épithélium alvéolaire. Les β2 agonistes intraveineux réduisent l’eau pulmonaire extravasculaire mais un essai randomisé a montré un excès de mortalité dans le groupe traité. Par voie inhalée, les β2 agonistes n’ont pas d’effet sur la survie (Gao Smith, et al. 2012, National Heart, et al. 2011) (Tableau 4).
Le monoxyde d’azote inhalé est un gaz qui provoque une relaxation de la fibre musculaire lisse vasculaire des vaisseaux des zones pulmonaires bien ventilées et tend à homogénéiser les rapports ventilation/perfusion pulmonaires. Une amélioration de l’oxygénation et une diminution de la pression artérielle pulmonaire peuvent en résulter.
Biothérapies et thérapeutiques immunomodulatrices
Les tableaux 5 et 6 listent les études principales ayant testé des biothérapies et autres thérapies immunomodulatrices dans le cadre thérapeutique du SDRA, ainsi que les études en cours, respectivement.
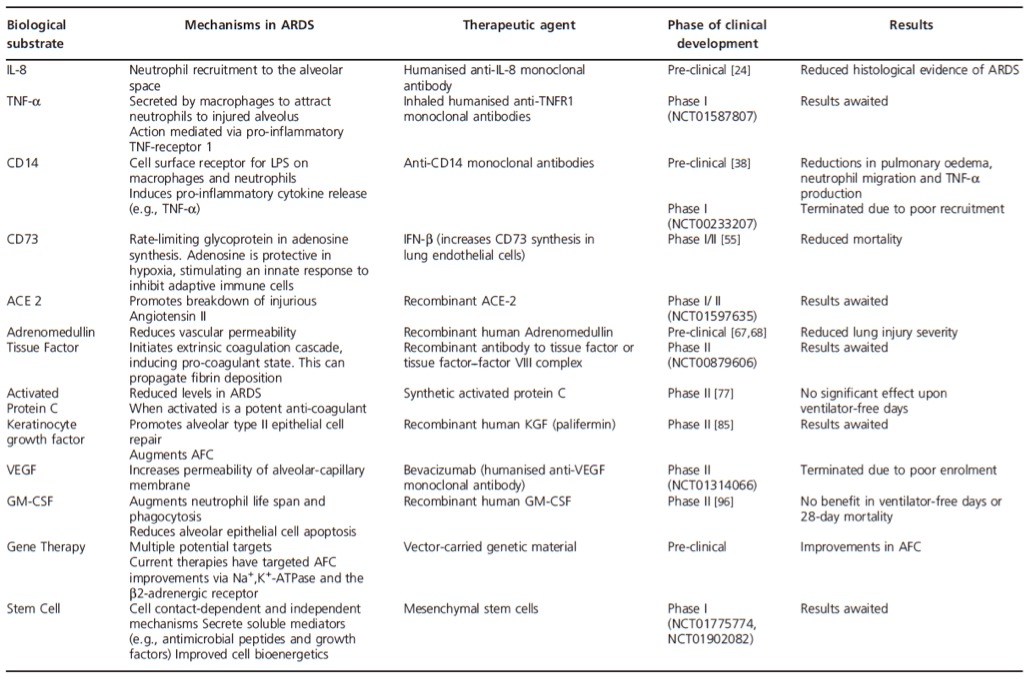
Tableau ‑5: Résumé des études ayant testé des biothérapies et des thérapies immunomodulatrices dans le SDRA. D’après (Boyle, et al. 2015)
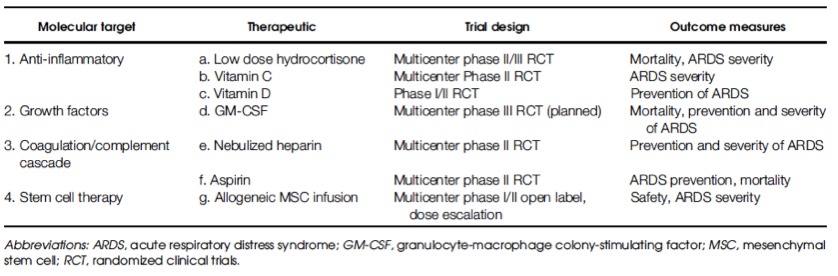
Tableau 6: Principales études en cours évaluant des thérapies immunomodulatrices dans le SDRA. D’après (Standiford and Ward 2016)
Les études ayant testé les propriétés pléiotropes et immunomodulatrices des statines dans le SDRA n’ont à ce jour démontré aucun bénéfice à leur usage préventif ou thérapeutique (National Heart, et al. 2014) (Xiong, et al. 2016).
Dans une étude prospective de cohorte de patients, un effet protecteur de l’aspirine en terme de réduction de mortalité a été démontré (Boyle, et al. 2015). Une étude multicentrique randomisée contrôlée est en cours depuis 2014 (NCT02326350). Les résultats ne sont pas encore connus.
En revanche, malgré de nombreux modèles expérimentaux ayant clairement démontré des effets thérapeutiques multiples du keratinocyte growth factor (KGF), la dernière étude multicentrique randomisée n’a retrouvé aucune différence en terme de morbi-mortalité dans le groupe des patients ayant reçu le traitement (Cross, et al. 2013).
Les autres biothérapies ou traitements immunomodulateurs listés dans les Tableaux 5 et 6 ont soit échoué, soit restent en cours d’investigations sans résultats définitifs.
L’échec de la plupart des biothérapies, pourtant tous préalablement jugés comme prometteurs à partir des données expérimentales, renouvelle la question qui avait été posé face aux nombreux échecs rencontrés dans les autres thérapeutiques évoquées plus haut. L’hétérogénéité du syndrome explique très probablement en partie la perte d’efficacité de ces thérapeutiques, une fois le pas de la translation vers la clinique franchi. Les modèles expérimentaux répliquent en effet des modèles très homogènes et stéréotypés. Les différents phénotypes rencontrés en clinique sont beaucoup plus hétérogènes, présentant des pronostics, des scores de gravité différents, et des réponses aux thérapeutiques probablement divergentes. La deuxième raison de ces échecs réside probablement dans le fait que mise à part les corticoïdes et les statines, les molécules testées ciblent une voie de signalisation bien précise. Or, nous l’avons vu précédemment, les voies de signalisation activées dans le SDRA sont multiples. Il semble illusoire, voire risqué, de modifier le devenir des patients atteints de SDRA via l’administration d’un agent pharmacologique n’agissant que sur l’une de ces voies. La biothérapie idéale serait un agent capable d’interagir avec la plupart des acteurs cellulaires impliqués dans la physiopathologie du SDRA, afin de moduler leur activité.
7. Références :
Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. The Acute Respiratory Distress Syndrome Network. N Engl J Med 2000; 342:1301-8.
Amato MB, Barbas CS, Medeiros DM, Magaldi RB, Schettino GP, Lorenzi-Filho G, Kairalla RA, Deheinzelin D, Munoz C, Oliveira R, Takagaki TY, Carvalho CR. Effect of a protective-ventilation strategy on mortality in the acute respiratory distress syndrome. N Engl J Med 1998; 338:347-54.
Ashbaugh DG, Bigelow DB, Petty TL, Levine BE. Acute respiratory distress in adults. Lancet 1967; 2:319-23.
Bernard GR, Artigas A, Brigham KL, Carlet J, Falke K, Hudson L, Lamy M, Legall JR, Morris A, Spragg R. The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am J Respir Crit Care Med 1994; 149:818-24.
Bernard GR, Luce JM, Sprung CL, Rinaldo JE, Tate RM, Sibbald WJ, Kariman K, Higgins S, Bradley R, Metz CA, et al. High-dose corticosteroids in patients with the adult respiratory distress syndrome. N Engl J Med 1987; 317:1565-70.
Bersten AD, Edibam C, Hunt T, Moran J, Australian, New Zealand Intensive Care Society Clinical Trials G. Incidence and mortality of acute lung injury and the acute respiratory distress syndrome in three Australian States. Am J Respir Crit Care Med 2002; 165:443-8.
Blondonnet R, Constantin JM, Sapin V, Jabaudon M. A Pathophysiologic Approach to Biomarkers in Acute Respiratory Distress Syndrome. Dis Markers 2016; 2016:3501373.
Boyle AJ, Di Gangi S, Hamid UI, Mottram LJ, McNamee L, White G, Cross LJ, McNamee JJ, O’Kane CM, McAuley DF. Aspirin therapy in patients with acute respiratory distress syndrome (ARDS) is associated with reduced intensive care unit mortality: a prospective analysis. Crit Care 2015; 19:109.
Brun-Buisson C, Minelli C, Bertolini G, Brazzi L, Pimentel J, Lewandowski K, Bion J, Romand JA, Villar J, Thorsteinsson A, Damas P, Armaganidis A, Lemaire F, Group AS. Epidemiology and outcome of acute lung injury in European intensive care units. Results from the ALIVE study. Intensive Care Med 2004; 30:51-61.
Buregeya E, Fowler RA, Talmor DS, Twagirumugabe T, Kiviri W, Riviello ED. Acute respiratory distress syndrome in the global context. Glob Heart 2014; 9:289-95.
Caser EB, Zandonade E, Pereira E, Gama AM, Barbas CS. Impact of distinct definitions of acute lung injury on its incidence and outcomes in Brazilian ICUs: prospective evaluation of 7,133 patients*. Crit Care Med 2014; 42:574-82.
Cross LJ, O’Kane CM, McDowell C, Elborn JJ, Matthay MA, McAuley DF. Keratinocyte growth factor in acute lung injury to reduce pulmonary dysfunction–a randomised placebo-controlled trial (KARE): study protocol. Trials 2013; 14:51.
Esteban A, Anzueto A, Frutos F, Alia I, Brochard L, Stewart TE, Benito S, Epstein SK, Apezteguia C, Nightingale P, Arroliga AC, Tobin MJ, Mechanical Ventilation International Study G. Characteristics and outcomes in adult patients receiving mechanical ventilation: a 28-day international study. JAMA 2002; 287:345-55.
Force ADT, Ranieri VM, Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, Fan E, Camporota L, Slutsky AS. Acute respiratory distress syndrome: the Berlin Definition. JAMA 2012; 307:2526-33.
Gao Smith F, Perkins GD, Gates S, Young D, McAuley DF, Tunnicliffe W, Khan Z, Lamb SE, investigators B-s. Effect of intravenous beta-2 agonist treatment on clinical outcomes in acute respiratory distress syndrome (BALTI-2): a multicentre, randomised controlled trial. Lancet 2012; 379:229-35.
Guerin C, Reignier J, Richard JC, Beuret P, Gacouin A, Boulain T, Mercier E, Badet M, Mercat A, Baudin O, Clavel M, Chatellier D, Jaber S, Rosselli S, Mancebo J, Sirodot M, Hilbert G, Bengler C, Richecoeur J, Gainnier M, Bayle F, Bourdin G, Leray V, Girard R, Baboi L, Ayzac L, Group PS. Prone positioning in severe acute respiratory distress syndrome. N Engl J Med 2013; 368:2159-68.
Han S, Mallampalli RK. The acute respiratory distress syndrome: from mechanism to translation. J Immunol 2015; 194:855-60.
Herridge MS, Cheung AM, Tansey CM, Matte-Martyn A, Diaz-Granados N, Al-Saidi F, Cooper AB, Guest CB, Mazer CD, Mehta S, Stewart TE, Barr A, Cook D, Slutsky AS, Canadian Critical Care Trials G. One-year outcomes in survivors of the acute respiratory distress syndrome. N Engl J Med 2003; 348:683-93.
Herridge MS, Tansey CM, Matte A, Tomlinson G, Diaz-Granados N, Cooper A, Guest CB, Mazer CD, Mehta S, Stewart TE, Kudlow P, Cook D, Slutsky AS, Cheung AM, Canadian Critical Care Trials G. Functional disability 5 years after acute respiratory distress syndrome. N Engl J Med 2011; 364:1293-304.
Hughes M, MacKirdy FN, Ross J, Norrie J, Grant IS, Scottish Intensive Care S. Acute respiratory distress syndrome: an audit of incidence and outcome in Scottish intensive care units. Anaesthesia 2003; 58:838-45.
Kuebler WM, Ying X, Singh B, Issekutz AC, Bhattacharya J. Pressure is proinflammatory in lung venular capillaries. J Clin Invest 1999; 104:495-502.
Li G, Malinchoc M, Cartin-Ceba R, Venkata CV, Kor DJ, Peters SG, Hubmayr RD, Gajic O. Eight-year trend of acute respiratory distress syndrome: a population-based study in Olmsted County, Minnesota. Am J Respir Crit Care Med 2011; 183:59-66.
Luhr OR, Antonsen K, Karlsson M, Aardal S, Thorsteinsson A, Frostell CG, Bonde J. Incidence and mortality after acute respiratory failure and acute respiratory distress syndrome in Sweden, Denmark, and Iceland. The ARF Study Group. Am J Respir Crit Care Med 1999; 159:1849-61.
Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. J Clin Invest 2012; 122:2731-40.
Meduri GU, Golden E, Freire AX, Taylor E, Zaman M, Carson SJ, Gibson M, Umberger R. Methylprednisolone infusion in early severe ARDS: results of a randomized controlled trial. Chest 2007; 131:954-63.
Meduri GU, Headley AS, Golden E, Carson SJ, Umberger RA, Kelso T, Tolley EA. Effect of prolonged methylprednisolone therapy in unresolving acute respiratory distress syndrome: a randomized controlled trial. JAMA 1998; 280:159-65.
Monsel A, Calfee CS. Focusing on the alveolar epithelium: Alveolar fluid clearance in diffuse versus focal acute respiratory distress syndrome. Anaesth Crit Care Pain Med 2016; 35:75-7.
Monsel A, Zhu YG, Gennai S, Hao Q, Liu J, Lee JW. Cell-based therapy for acute organ injury: preclinical evidence and ongoing clinical trials using mesenchymal stem cells. Anesthesiology 2014; 121:1099-121.
National Heart L, Blood Institute ACTN, Truwit JD, Bernard GR, Steingrub J, Matthay MA, Liu KD, Albertson TE, Brower RG, Shanholtz C, Rock P, Douglas IS, deBoisblanc BP, Hough CL, Hite RD, Thompson BT. Rosuvastatin for sepsis-associated acute respiratory distress syndrome. N Engl J Med 2014; 370:2191-200.
National Heart L, Blood Institute Acute Respiratory Distress Syndrome Clinical Trials N, Matthay MA, Brower RG, Carson S, Douglas IS, Eisner M, Hite D, Holets S, Kallet RH, Liu KD, MacIntyre N, Moss M, Schoenfeld D, Steingrub J, Thompson BT. Randomized, placebo-controlled clinical trial of an aerosolized beta(2)-agonist for treatment of acute lung injury. Am J Respir Crit Care Med 2011; 184:561-8.
National Heart L, Blood Institute Acute Respiratory Distress Syndrome Clinical Trials N, Wheeler AP, Bernard GR, Thompson BT, Schoenfeld D, Wiedemann HP, deBoisblanc B, Connors AF, Jr., Hite RD, Harabin AL. Pulmonary-artery versus central venous catheter to guide treatment of acute lung injury. N Engl J Med 2006; 354:2213-24.
National Heart L, Blood Institute Acute Respiratory Distress Syndrome Clinical Trials N, Wiedemann HP, Wheeler AP, Bernard GR, Thompson BT, Hayden D, deBoisblanc B, Connors AF, Jr., Hite RD, Harabin AL. Comparison of two fluid-management strategies in acute lung injury. N Engl J Med 2006; 354:2564-75.
Ortega-Gomez A, Perretti M, Soehnlein O. Resolution of inflammation: an integrated view. EMBO Mol Med 2013; 5:661-74.
Papazian L, Forel JM, Gacouin A, Penot-Ragon C, Perrin G, Loundou A, Jaber S, Arnal JM, Perez D, Seghboyan JM, Constantin JM, Courant P, Lefrant JY, Guerin C, Prat G, Morange S, Roch A, Investigators AS. Neuromuscular blockers in early acute respiratory distress syndrome. N Engl J Med 2010; 363:1107-16.
Pierrakos C, Vincent JL. The changing pattern of acute respiratory distress syndrome over time: a comparison of two periods. Eur Respir J 2012; 40:589-95.
Reynolds HN, McCunn M, Borg U, Habashi N, Cottingham C, Bar-Lavi Y. Acute respiratory distress syndrome: estimated incidence and mortality rate in a 5 million-person population base. Crit Care 1998; 2:29-34.
Rogers AJ, Matthay MA. Applying metabolomics to uncover novel biology in ARDS. Am J Physiol Lung Cell Mol Physiol 2014; 306:L957-61.
Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, Stern EJ, Hudson LD. Incidence and outcomes of acute lung injury. N Engl J Med 2005; 353:1685-93.
Spragg RG, Lewis JF, Walmrath HD, Johannigman J, Bellingan G, Laterre PF, Witte MC, Richards GA, Rippin G, Rathgeb F, Hafner D, Taut FJ, Seeger W. Effect of recombinant surfactant protein C-based surfactant on the acute respiratory distress syndrome. N Engl J Med 2004; 351:884-92.
Standiford TJ, Ward PA. Therapeutic targeting of acute lung injury and acute respiratory distress syndrome. Transl Res 2016; 167:183-91.
Steinberg KP, Hudson LD, Goodman RB, Hough CL, Lanken PN, Hyzy R, Thompson BT, Ancukiewicz M, National Heart L, Blood Institute Acute Respiratory Distress Syndrome Clinical Trials N. Efficacy and safety of corticosteroids for persistent acute respiratory distress syndrome. N Engl J Med 2006; 354:1671-84.
Tecchio C, Micheletti A, Cassatella MA. Neutrophil-derived cytokines: facts beyond expression. Front Immunol 2014; 5:508.
Villar J, Kacmarek RM, Perez-Mendez L, Aguirre-Jaime A. A high positive end-expiratory pressure, low tidal volume ventilatory strategy improves outcome in persistent acute respiratory distress syndrome: a randomized, controlled trial. Crit Care Med 2006; 34:1311-8.
Ware LB. Pathophysiology of acute lung injury and the acute respiratory distress syndrome. Seminars in respiratory and critical care medicine 2006; 27:337-49.
Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med 2000; 342:1334-49.
Wind J, Versteegt J, Twisk J, van der Werf TS, Bindels AJ, Spijkstra JJ, Girbes AR, Groeneveld AB. Epidemiology of acute lung injury and acute respiratory distress syndrome in The Netherlands: a survey. Respir Med 2007; 101:2091-8.
Xiong B, Wang C, Tan J, Cao Y, Zou Y, Yao Y, Qian J, Rong S, Huang Y, Huang J. Statins for the prevention and treatment of acute lung injury and acute respiratory distress syndrome: a systematic review and meta-analysis. Respirology 2016.
